The Genetic Basis of Disease
Top Milestones in Human Genetics and Genomics Over 75 years (1948-2023)
Beginning in the 1950s, researchers first used a molecular technique called karyotyping to visualize human chromosomes and their differences linked to conditions such as Down syndrome. Much progress has been made since then. We have identified many other causes of genetic anomalies, including single nucleotide variants and larger structural changes that alter protein-encoding genes. We are now starting to understand the role of variation in regions that control the expression of genes and how that impacts human diseases. We are just now starting to understand the role of variation in non-protein coding parts of the genome and their role in human diseases.
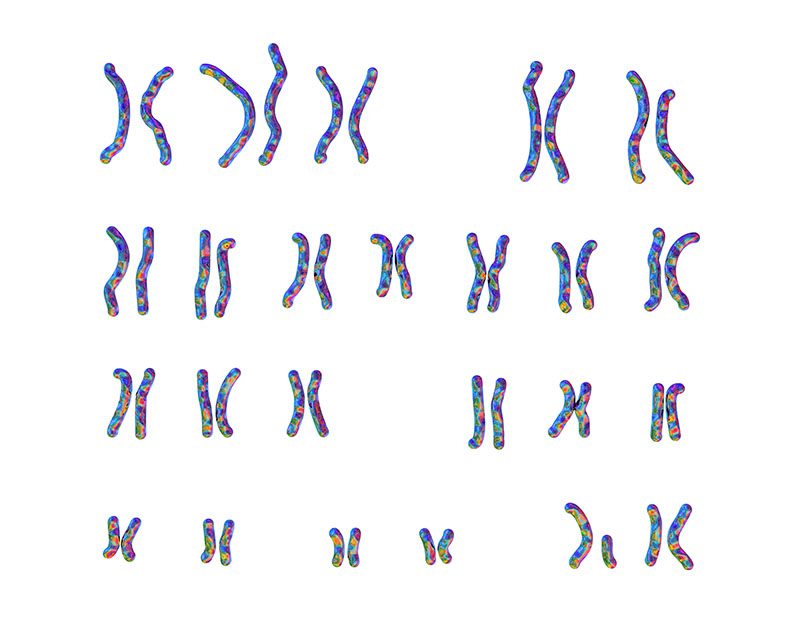
Example of Karyotype
1950s-present
Discovering anomalies in chromosomes, such as a missing copy of a chromosome or duplicate copies, was helped by the invention of karyotyping techniques that allow paired human chromosomes to be easily stained, arranged, and visualized. These techniques enabled the first examples of molecular characterization of human diseases. Chromosomal conditions such as Down syndrome, where an individual has an extra copy of chromosome 21, were initially discovered through karyotyping. DiscoverGenetics-FactSheet-AnalyzingDNA.pdf
Sources: Karyotyping | Learn Science at Scitable (nature.com); Karyotyping: Different Methods and Their Significance in Cell-Based Research (synthego.com); and History of Karyotyping and Cytogenetics – KaryotypingHub
1950s
The discovery of the molecular basis of single gene disorders, or those conditions linked to a specific genetic variant, was established when the variant that causes sickle cell disease was identified. The key to making this discovery was uncovering the amino acid change that causes a structural change in the hemoglobin protein, which carries oxygen in red blood cells.
Source: History of Sickle Cell Disease – Rare Disease Advisor
1980s
To further characterize single gene disorders, a series of molecular techniques known as positional cloning were developed. These techniques made it possible to uncover the gene responsible for a given condition and its relationship to the physical characteristics manifested by that condition.
Source: Teaching molecular genetics: chapter 4—positional cloning of genetic disorders – PMC (nih.gov)
1990s-2000s
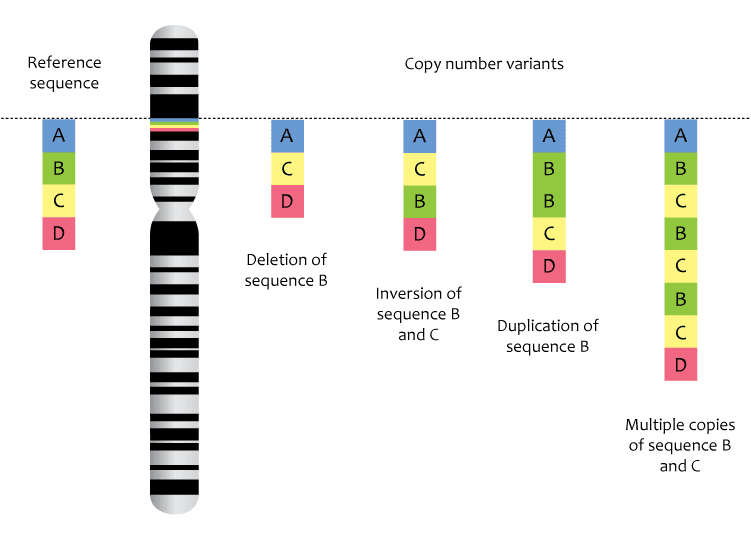
During this period, it became clear that some disorders were caused by structural changes in the genome. These changes often result from copy number variants (CNVs), which could result in duplicate copies of genes or a loss of a gene copy or a portion of the copy. Charcot-Marie-Tooth disease, which causes degeneration of the peripheral nerves, is an example of a CNV disorder for which a duplication of a specific region in chromosome 17 is the most common cause.
Sources: Gonzaga-Jauregui, Claudia. “The Human Genome.” In Brenner’s Encyclopedia of Genetics, 3rd Edition.; Genomic disorders ten years on – PMC (nih.gov); Charcot–Marie–Tooth Disease (CMT): Symptoms, Treatment, Facts (cmtausa.org)
2005
Understanding of haplotypes and the development of genotyping technologies led to the implementation of large-scale genome-wide association studies (GWAS), which allowed for the scanning of markers across the genomes of many people. This approach uncovered genetic markers associated with common diseases such as asthma, diabetes, cancer, and many others. It is also paving the way to personalized medicine, where treatments are modified based on an individual’s genetic profile.
2010-Present
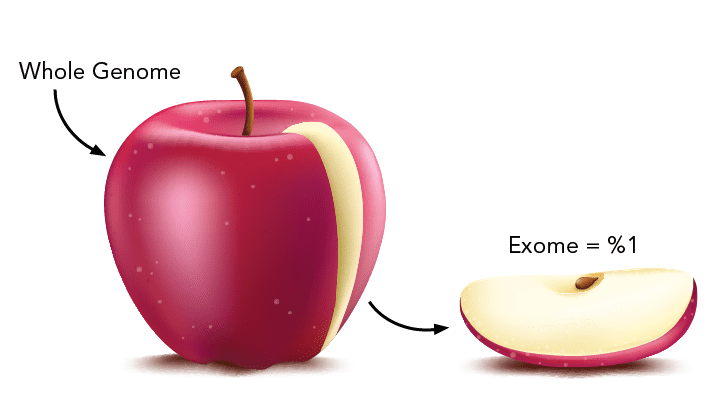
Genetic diseases are often caused by variants in DNA sequences associated with genes that encode proteins. This subsection, called the exome, makes up only 2% of the genome. Exome sequencing has proven to be an efficient and cost-effective way to discover the cause of highly penetrant genetic diseases. Reduction in the cost of sequencing over the last 13 years is facilitating the utilization of whole genome sequencing, which is increasingly being used to identify causal variation in non-coding regions outside the exome.